Dr. Eke is from the University Hospital, Birmingham, United Kingdom. Drs. Bailey and Gach are from University Hospital Coventry and Warwickshire NHS Trust, United Kingdom.
The authors report no conflict of interest.
Correspondence: Ure Eke, MBChB, MRCP, University Hospital Birmingham, Edgbaston, Birmingham B15 2TH, United Kingdom (ure_eke@hotmail.com).
A 12-year-old girl presented with an asymptomatic hypopigmented area on the right cheek of 2 months’ duration. Two years prior to presentation she was diagnosed with neurofibromatosis type I (NF1) based on the findings of 13 café au lait spots on the trunk, axillary and groin freckling, bilateral Lisch nodules, and mild scoliosis. She was otherwise well and had no relevant medical history or family history of neurofibromatosis.
A 12-year-old girl presented with an asymptomatic hypopigmented area on the right cheek of 2 months’ duration. Two years prior to presentation she was diagnosed with neurofibromatosis type I (NF1) based on the findings of 13 café au lait spots on the trunk, axillary and groin freckling, bilateral Lisch nodules, and mild scoliosis. She was otherwise well and had no relevant medical history or family history of neurofibromatosis.
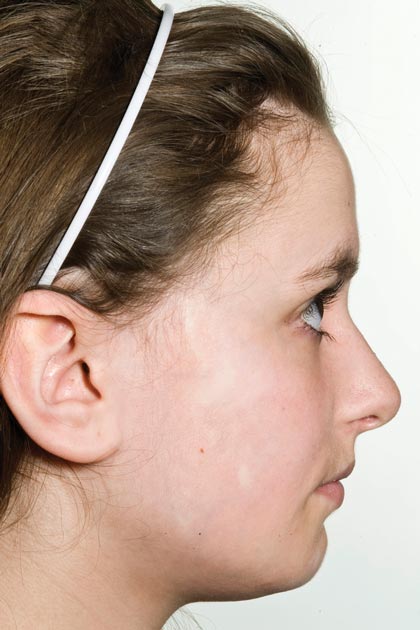
Physical examination revealed a 3×7-cm linear, shiny, sclerotic plaque extending from the right temple to the preauricular area and lower aspect of the right cheek (Figure 1) with associated facial asymmetry (Figure 2). A smaller similar plaque on the chin measured 0.5×0.5 cm. Examination of the oral cavity was unremarkable and there were no neurological signs. No other features were present to suggest a mixed connective tissue disease or lupus erythematosus, and nuclear antibodies were negative.
Figure 1. Linear sclerotic plaque extending from the right temple to the right preauricular area and right cheek.
Figure 2. Facial asymmetry with contraction of the right cheek.
An incisional biopsy from the sclerotic plaque revealed swollen eosinophilic bundles in the reticular dermis with a moderate perivascular lymphohistiocytic infiltrate extending into the subcutaneous layer. These features were compatible with the diagnosis of linear scleroderma. Due to the progressive nature of the condition and its anatomical location, she was treated with pulsed intravenous methylprednisolone 1 g daily for 3 days and commenced on oral methotrexate 20 mg weekly as well as tacrolimus ointment 0.1% daily. The plaque gradually softened and faded over a period of 8 months. The patient continued on methotrexate for another 10 months. The facial asymmetry persisted with a discernible reduction in the volume of the right cheek. New onset of ipsilateral jaw locking and pain associated with spasms of the muscles of mastication suggested the diagnosis of Parry-Romberg syndrome.
Neurofibromatosis type I is a neuroectodermal abnormality first described by Friedrich von Recklinghausen in 1882 with an incidence of 1 in 3000 births. Neurofibromatosis type I gene mutations lead to increased Ras activity, which is implicated in many NF1-related conditions such as neurofibromas and schwannomas.1 Autoimmune conditions including systemic lupus erythematosus (SLE) and mixed connective tissue rarely have been reported in NF1, but the mechanism of their association is not clear.2 Based on a review of 5 reported cases of NF1 and SLE, most patients were female, and the predominant features of NF1 were café au lait macules and neurofibromas.3-6 One case documented a family history of NF1,6 suggesting predominance of sporadic mutations in these cases. Interestingly, in 3 cases the diagnosis of SLE preceded the diagnosis of NF1, prompting the authors to suggest a viral trigger for the development of NF1 lesions.3,4 Linear scleroderma is immunologically mediated and is characterized by the onset of smooth indurated cutaneous plaques. According to a PubMed search of articles indexed for MEDLINE using the search terms morphea and neurofibromatosis as well as linear scleroderma and neurofibromatosis, there have been no reports of linear scleroderma or morphea associated with NF1. Cichowski et al7 demonstrated enhanced activation of Ras and prolonged activities of both Ras and extracellular signal-regulated kinase (ERK) signaling pathways in NF1-deficient mice. Chen et al8 showed that heparin sulfate–dependent ERK activation contributes to the development of scleroderma by promoting the expression of profibrotic proteins in scleroderma fibroblasts. It was previously noted that increased Ras/ERK signaling activities were important in connective tissue growth factor expression in normal mesenchymal cells.9
Based on these findings, we speculated that hyperactivation of Ras/ERK signaling from NF1 mutations could lead to the promotion of fibrosis seen in scleroderma. The lack of similar reports, however, suggests that the presence of both conditions in this case is coincidental. However, the growing number of reports on autoimmune and connective tissue disorders in NF1 reflects the need for further research in this area.